HEPSERA™ (Gilead)
adefovir dipivoxil Tablets
Rx Only
WARNINGS
- SEVERE ACUTE EXACERBATIONS OF HEPATITIS HAVE BEEN REPORTED IN PATIENTS WHO HAVE DISCONTINUED ANTI-HEPATITIS B THERAPY, INCLUDING THERAPY WITH HEPSERA. HEPATIC FUNCTION SHOULD BE MONITORED CLOSELY IN PATIENTS WHO DISCONTINUE ANTI-HEPATITIS B THERAPY. IF APPROPRIATE, RESUMPTION OF ANTI-HEPATITIS B THERAPY MAY BE WARRANTED (SEE WARNINGS).
- IN PATIENTS AT RISK OF OR HAVING UNDERLYING RENAL DYSFUNCTION, CHRONIC ADMINISTRATION OF HEPSERA MAY RESULT IN NEPHROTOXICITY. THESE PATIENTS SHOULD BE MONITORED CLOSELY FOR RENAL FUNCTION AND MAY REQUIRE DOSE ADJUSTMENT (SEE WARNINGS AND DOSAGE AND ADMINISTRATION).
- HIV RESISTANCE MAY EMERGE IN CHRONIC HEPATITIS B PATIENTS WITH UNRECOGNIZED OR UNTREATED HUMAN IMMUNODEFICIENCY VIRUS (HIV) INFECTION TREATED WITH ANTI-HEPATITIS B THERAPIES, SUCH AS THERAPY WITH HEPSERA, THAT MAY HAVE ACTIVITY AGAINST HIV (SEE WARNINGS).
- LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS, INCLUDING FATAL CASES, HAVE BEEN REPORTED WITH THE USE OF NUCLEOSIDE ANALOGS ALONE OR IN COMBINATION WITH OTHER ANTIRETROVIRALS (SEE WARNINGS).
DESCRIPTION
HEPSERA is the tradename for adefovir dipivoxil, a diester prodrug of adefovir. Adefovir is an acyclic nucleotide analog with activity against human hepatitis B virus (HBV).
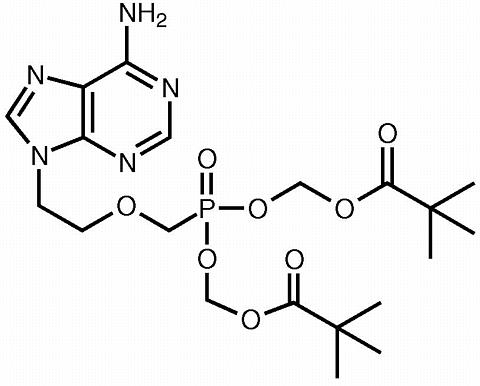
The chemical name of adefovir dipivoxil is 9-[2-[bis[(pivaloyloxy)methoxy]phosphinyl]methoxy]ethyl]adenine. It has a molecular formula of C 20 H 32 N 5 O 8 P, a molecular weight of 501.48 and the following structural formula:
 Adefovir dipivoxil is a white to off-white crystalline powder with an
aqueous solubility of 19 mg/mL at pH 2.0 and 0.4 mg/mL at pH 7.2. It has
an octanol/aqueous phosphate buffer (pH 7) partition coefficient (log
p) of 1.91.
Adefovir dipivoxil is a white to off-white crystalline powder with an
aqueous solubility of 19 mg/mL at pH 2.0 and 0.4 mg/mL at pH 7.2. It has
an octanol/aqueous phosphate buffer (pH 7) partition coefficient (log
p) of 1.91.
HEPSERA tablets are for oral administration. Each tablet contains 10 mg of adefovir dipivoxil and the following inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, pregelatinized starch, and talc.
Microbiology
Mechanism of Action:
Adefovir is an acyclic nucleotide analog of adenosine monophosphate. Adefovir is phosphorylated to the active metabolite, adefovir diphosphate, by cellular kinases. Adefovir diphosphate inhibits HBV DNA polymerase (reverse transciptase) by competing with the natural substrate deoxyadenosine triphosphate and by causing DNA chain termination after its incorporation into viral DNA. The inhibition constant (K i ) for adefovir diphosphate for HBV DAN polymerase was 0.1 µM. Adefovir diphosphate is a weak inhibitor of human DNA polymerases (alpha) and (gamma) with K i values of 1.18 µM and 0.97 µM, respectively.
Antiviral Activity:
The in vitro antiviral activity of adefovir was determined in HBV transfected human hepatoma cell lines. The concentration of adefovir that inhibited 50% of viral DNA synthesis (IC 50 ) varied from 0.2 to 2.5 µM.
Drug Resistance:
Clinical Studies 437 & 438
Genotypic and phenotypic analyses of serum HBV DNA from adefovir dipivoxil (10 mg or 30 mg) treated HBeAg-positive patients (n = 215; study 437) and HBeAg-negative patients (n = 56; study 438) at baseline and week 48 did not identify mutations in the HBV DNA polymerase gene that may confer reduced susceptibility to adefovir. An unconfirmed increase of >/= 1 log 10 copies/mL in serum HBV DNA was observed in some patients. The molecular basis and/or the clinical significance for the observed unconfirmed increases are not known.
Cross-resistance:
Recombinant HBV variants containing lamivudine-resistance-associated mutations (L528M, M552I, M552V, L528M + M552V) in the HBV DNA polymerase gene were susceptible to adefovir in vitro . Adefovir has also demonstrated anti-HBV activity (median reduction in serum HBV DNA of 4.3 log 10 copies/mL) against clinical isolates of HBV containing lamivudine-resistance-associated mutations (study 435). HBV variants with DNA polymerase mutations T476N and R or W501Q associated with resistance to hepatitis B immunoglobulin were susceptible to adefovir in vitro .
CLINICAL PHARMACOLOGY
Pharmacokinetics
The pharmacokinetics of adefovir have been evaluated in healthy volunteers and patients with chronic hepatitis B. Adefovir pharmacokinetics are similar between these populations.
Absorption:
Adefovir dipivoxil is a diester prodrug of the active moiety adefovir. Based on a cross study comparison, the approximate oral bioavailability of adefovir from a 10 mg single dose of HEPSERA is 59%.
Following oral administration of a 10 mg single dose of HEPSERA to chronic hepatitis B patients (n = 14), the peak adefovir plasma concentration (C max ) was 18.4 ± 6.26 ng/mL (mean ± SD) and occurred between 0.58 and 4.00 hours (median = 1.75 hours) post dose. The adefovir area under the plasma concentration-time curve (AUC 0-(infinity) ) was 220 ± 70.0 ng•h/mL. Plasma adefovir concentrations declined in a biexponential manner with a terminal elimination half-life of 7.48 ± 1.65 hours.
The pharmacokinetics of adefovir in subjects with adequate renal function were not affected by once daily dosing of 10 mg HEPSERA over seven days. The impact of long-term once daily administration of 10 mg HEPSERA on adefovir pharmacokinetics has not been evaluated.
Effects of Food on Oral Absorption:
Adefovir exposure was unaffected when a 10 mg single dose of HEPSERA was administered with food (an approximately 1000 kcal high-fat meal). HEPSERA may be taken without regard to food.
Distribution:
In vitro binding of adefovir to human plasma or human serum proteins is </= 4% over the adefovir concentration range of 0.1 to 25 µg/mL. The volume of distribution at steady-state following intravenous administration of 1.0 or 3.0 mg/kg/day is 392 ± 75 and 352 ± 9 mL/kg, respectively.
Metabolism and Elimination:
Following oral administration, adefovir dipivoxil is rapidly converted to adefovir. Forty-five percent of the dose is recovered as adefovir in the urine over 24 hours at steady-state following 10 mg oral doses of HEPSERA. Adefovir is renally excreted by a combination of glomerular filtration and active tubular secretion ( See Drug Interactions ).
Special Populations:
Gender
The pharmacokinetics of adefovir were similar in male and female patients.
Race
Insufficient data are available to determine the effect of race on the pharmacokinetics of adefovir.
Pediatric and Geriatric Patients
Pharmacokinetic studies have not been conducted in children or in the elderly.
Renal Impairment
In subjects with moderately or severely impaired renal function or with end-stage renal disease (ESRD) requiring hemodialysis, C max , AUC, and half-life (T 1/2 ) were increased compared to subjects with normal renal function. It is recommended that the dosing interval of HEPSERA be modified in these patients (See DOSAGE AND ADMINISTRATION ).
The pharmacokinetics of adefovir in non-chronic hepatitis B patients with varying degrees of renal impairment are described in Table 1. In this study, subjects received a 10 mg single dose of HEPSERA.
Table 1. Pharmacokinetic Parameters (Mean ± SD) of Adefovir in Patients with Varying Degrees of Renal Function
| Renal Function Group | Unimpaired | Mild | Moderate | Severe |
|---|---|---|---|---|
| Baseline Creatinine Clearance (mL/min) | > 80 (n = 7) |
50 - 80 (n = 8) |
30 - 49 (n = 7) |
10 - 29 (n = 10) |
| C max (ng/mL) | 17.8 ± 3.22 | 22.4 ± 4.04 | 28.5 ± 8.57 | 51.6 ± 10.3 |
| AUC 0-(infinity) (ng•h/mL) | 201 ± 40.8 | 266 ± 55.7 | 455 ± 176 | 1240 ± 629 |
| CL/F (mL/min) | 469 ± 99.0 | 356 ± 85.6 | 237 ± 118 | 91.7 ± 51.3 |
| CL renal (mL/min) | 231 ± 48.9 | 148 ± 39.3 | 83.9 ± 27.5 | 37.0 ± 18.4 |
A four-hour period of hemodialysis removed approximately 35% of the adefovir dose. The effect of peritoneal dialysis on adefovir removal has not been evaluated.
Hepatic Impairment
The pharmacokinetics of adefovir following a 10 mg single dose of HEPSERA have been studied in non-chronic hepatitis B patients with hepatic impairment. There were no substantial alterations in adefovir pharmacokinetics in patients with moderate and severe hepatic impairment compared to unimpaired patients. No change in HEPSERA dosing is required in patients with hepatic impairment.
Drug Interactions:
Adefovir dipivoxil is rapidly converted to adefovir in vivo . At concentrations substantially higher (> 4000 fold) than those observed in vivo , adefovir did not inhibit any of the common human CYP450 enzymes, CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Adefovir is not a substrate for these enzymes. However, the potential for adefovir to induce CYP450 enzymes is unknown. Based on the results of these in vitro experiments and the renal elimination pathway of adefovir, the potential for CYP450 mediated interactions involving adefovir as an inhibitor or substrate with other medicinal products is low.
The pharmacokinetics of adefovir have been evaluated following multiple dose administration of HEPSERA (10 mg once daily) in combination with lamivudine (100 mg once daily), trimethoprim/sulfamethoxazole (160/800 mg twice daily), acetaminophen (1000 mg four times daily) and ibuprofen (800 mg three times daily) in healthy volunteers (n = 18 per study).
Adefovir did not alter the pharmacokinetics of lamivudine, trimethoprim/sulfamethoxazole, acetaminophen and ibuprofen.
The pharmacokinetics of adefovir were unchanged when HEPSERA was co-administered with lamivudine, trimethoprim/sulfamethoxazole and acetaminophen. When HEPSERA was co-administered with ibuprofen (800 mg three times daily) increases in adefovir C max (33%), AUC (23%) and urinary recovery were observed. This increase appears to be due to higher oral bioavailability, not a reduction in renal clearance of adefovir.
INDICATIONS AND USAGE
HEPSERA is indicated for the treatment of chronic hepatitis B in adults with evidence of active viral replication and either evidence of persistent elevations in serum aminotransferases (ALT or AST) or histologically active disease.
This indication is based on histological, virological, biochemical, and serological responses in adult patients with HBeAg+ and HBeAg- chronic hepatitis B with compensated liver function, and in adult patients with clinical evidence of lamivudine-resistant hepatitis B virus with either compensated or decompensated liver function.
Description of Clinical Studies
HBeAg-positive Chronic Hepatitis B:
Study 437 was a randomized, double-blind, placebo-controlled, three-arm study in patients with HBeAg-positive chronic hepatitis B that allowed for a comparison between placebo and HEPSERA. The median age of patients was 33 years. Seventy-four percent were male, 59% were Asian, 36% were Caucasian and 24% has prior interferon-(alpha) treatment. At baseline, patients had a median total Knodell Histology Activity Index (HAI) score of 10, a median serum HBV DNA level as measured by an experimental polymerase chain reaction assay of 8.36 log 10 copies/mL and a median ALT level of 2.3 times the upper limit of normal.
HBeAg-negative (anti-HBe positive/HBV DNA positive) Chronic Hepatitis B:
Study 438 was a randomized, double-blind, placebo-controlled study in patients who were HBeAg-negative at screening, and anti-HBe positive. The median age of patients was 46 years. Eight-three percent were male, 66% were Caucasian, 30% were Asian and 41% had prior interferon-(alpha) treatment. At baseline, the median total Knodell HAI score was 10, the median serum HBV DNA level as measured by an experimental polymerase chain reaction assay was 7.08 log 10 copies/mL, and the median ALT was 2.3 times the upper limit of normal.
The primary efficacy endpoint in both studies was histological improvement at week 48; results of which are shown in Table 2.
Table 2. Histological Response at Week 48 *
| Study 437 | Study 438 | |||
| HEPSERA 10 mg |
Placebo | HEPSERA 10 mg |
Placebo | |
| (n = 168) | (n = 161) | (n = 121) | (n = 57) | |
| Improvement ** | 53% | 25% | 64% | 35% |
| No Improvement | 37% | 67% | 29% | 63% |
| Missing/Unassessable Data | 10% | 7% | 7% | 2% |
*Intent-to-Treat population (patients with >/= 1 dose of study drug) with assessable baseline biopsies
**Histological improvement defined as >/= 2 point decrease in the Knodell necro-inflammatory score with no worsening of the Knodell fibrosis score
Table 3 illustrates the changes in Ishak Fibrosis Score by treatment group.
Table 3. Changes in Ishak Fibrosis Score at Week 48
| Study 437 | Study 438 | |||
| HEPSERA 10 mg |
Placebo | HEPSERA 10 mg |
Placebo | |
| Number of adequate biopsy pairs | (n = 150) | (n = 146) | (n = 112) | (n = 55) |
| Ishak Fibrosis Score Improved * | 34% | 19% | 34% | 14% |
| Unchanged | 55% | 60% | 62% | 50% |
| Worsened | 11% | 21% | 4% | 36% |
*Change of 1 point or more in Ishak Fibrosis Score
At week 48, improvement was seen in respect to mean change in serum HBV
DNA (log 10 copies/mL), normalization of ALT, and HBeAg seroconversion
as compared to placebo in patients receiving HEPSERA (Table 4).
Table 4. Change in Serum HBV DNA, ALT Normalization, and HBeAg Seroconversion at Week 48
| Study 437 | Study 438 | |||
| HEPSERA 10 mg |
Placebo | HEPSERA 10 mg |
Placebo | |
| (n = 167) | (n = 171) | (n = 123) | (n = 61) | |
| Mean change ± SD in serum HBV DNA from baseline (log 10 copies/mL) |
-3.57 ± 1.64 |
-0.98 ± 1.32 | -3.65 ± 1.14 | -1.32 ± 1.25 |
| ALT Normalization | 48% | 16% | 72% | 29% |
| HBeAg Seroconversion | 12% | 6% | NA * | NA * |
*Patients with HBeAg-negative disease cannot undergo HBeAg seroconversion
In studies 437 and 438, continued treatment with HEPSERA to 72 weeks resulted in continued maintenance of mean reductions in serum HBV DNA observed at week 48. An increase in the proportion of patients with ALT normalization was also observed in study 437. The effect of continued treatment with HEPSERA on seroconversion is unknown.
Pre- and Post-Liver Transplantation Patients:
HEPSERA was also evaluated in an open-label, uncontrolled study of 324 chronic hepatitis B patients pre- (n = 128) and post- (n = 196) liver transplantation with clinical evidence of lamivudine-resistant hepatitis B virus (study 435). The median baseline HBV DNA as measured by an experimental polymerase chain reaction assay was 7.4 and 8.2 log 10 copies/mL, and the median baseline ALT was 1.8 and 2.1 times the upper limit of normal in pre- and post-liver transplantation patients, respectively. Results of this study are displayed in Table 5. Treatment with HEPSERA resulted in a similar reduction in serum HBV DNA regardless of the patterns of lamivudine-resistant HBV DNA polymerase mutations at baseline. The clinical significance of these findings as they relate to histological improvement is not known.
Table 5. Efficacy in Pre- and Post-Liver Transplantation Patients at Week 48
| Efficacy Parameter | Pre-Liver Transplantation | Post-Liver Transplantation |
| (n = 128) | (n = 196) | |
| Mean change ± SD in serum HBV DNA from baseline (log 10 copies/mL) | -3.8 ± 1.4 | -4.1 ± 1.6 |
| Stable or improved Child-Pugh-Turcotte score | 92% * | 96% |
| Normalization of: ** | ||
| ALT | 76% | 49% |
| Albumin | 81% | 76% |
| Bilirubin | 50% | 75% |
| Prothrombin time | 83% | 20% |
*24 week data
**Denominator in patients with abnormal values at baseline
Clinical Evidence of Lamivudine Resistance:
In study 461, an ongoing double-blind, active-controlled study in 59 chronic hepatitis B patients with clinical evidence of lamivudine-resistant hepatitis B virus, patients were randomized to receive either HEPSERA monotherapy or HEPSERA in combination with lamivudine 100 mg or lamivudine 100 mg alone. At week 16, the mean ± SD decrease in serum HBV DNA as measured by an experimental polymerase chain reaction assay was 3.11 ± 0.94 log 10 copies/mL for patients treated with HEPSERA and 2.95 ± 0.64 log 10 copies/mL for patients treated with HEPSERA in combination with lamivudine. There was a mean decrease in serum HBV DNA of 0.00 ± 0.28 log 10 copies/mL in patients receiving lamivudine alone. The clinical significance of these observed changes in serum HBV DNA has not yet been established.
CONTRAINDICATIONS
HEPSERA is contraindicated in patients with previously demonstrated hypersensitivity to any of the components of the product.
WARNINGS
Exacerbations of Hepatitis after Discontinuation of Treatment
Severe acute exacerbation of hepatitis has been reported in patients who have discontinued anti-hepatitis B therapy, including therapy with HEPSERA. Patients who discontinue HEPSERA should be monitored at repeated intervals over a period of time for hepatic function. If appropriate, resumption of anti-hepatitis B therapy may be warranted.
In clinical trials of HEPSERA, exacerbations of hepatitis (ALT elevations 10 times the upper limit of normal or greater) occurred in up to 25% of patients after discontinuation of HEPSERA. Most of these events occurred within 12 weeks of drug discontinuation. These exacerbations generally occurred in the absence of HBeAg seroconversion, and presented as serum ALT elevations in addition to re-emergence of viral replication. In the HBeAg-positive and HBeAg-negative studies in patients with compensated liver function, the exacerbations were not generally accompanied by hepatic decompensation. However, patients with advanced liver disease or cirrhosis may be at higher risk for hepatic decompensation. Although most events appear to have been self-limited or resolved with re-initiation of treatment, severe hepatitis exacerbations, including fatalities, have been reported. Therefore, patients should be closely monitored after stopping treatment.
Nephrotoxicity
Nephrotoxicity characterized by a delayed onset of gradual increases in serum creatinine and decreases in serum phosphorus was historically shown to be the treatment-limiting toxicity of adefovir dipivoxil therapy at substantially higher doses in HIV-infected patients (60 and 120 mg daily) and in chronic hepatitis B patients (30 mg daily). Chronic administration of HEPSERA (10 mg once daily) may result in nephrotoxicity. The overall risk of nephrotoxicity in patients with adequate renal function is low. However, this is of special importance in patients at risk of or having underlying renal dysfunction and patients taking concomitant nephrotoxic agents such as cyclosporine, tacrolimus, aminoglycosides, vancomycin and non-steroidal anti-inflammatory drugs (See ADVERSE REACTIONS ).
It is important to monitor renal function for all patients during treatment with HEPSERA, particularly for those with pre-existing or other risks for renal impairment. Patients with renal insufficiency at baseline or during treatment may require dose adjustment (See DOSAGE AND ADMINISTRATION ). The risks and benefits of HEPSERA treatment should be carefully evaluated prior to discontinuing HEPSERA in a patient with treatment-emergent nephrotoxicity.
HIV Resistance
Prior to initiating HEPSERA therapy, HIV antibody testing should be offered to all patients. Treatment with anti-hepatitis B therapies, such as HEPSERA, that have activity against HIV in a chronic hepatitis B patient with unrecognized or untreated HIV infection may result in emergence of HIV resistance. HEPSERA has not been shown to suppress HIV RNA in patients; however, there are limited data on the use of HEPSERA to treat patients with chronic hepatitis B co-infected with HIV.
Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs alone or in combination with antiretrovirals.
A majority of these cases have been in women. Obesity and prolonged nucleoside exposure may be risk factors. Particular caution should be exercised when administering nucleoside analogs to any patient with known risk factors for liver disease; however, cases have also been reported in patients with no known risk factors. Treatment with HEPSERA should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
PRECAUTIONS
Drug Interactions
Since adefovir is eliminated by the kidney, co-administration of HEPSERA with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of either adefovir and/or these co-administered drugs.
Apart from lamivudine, trimethoprim/sulfamethoxazole and acetaminophen, the effects of co-administration of HEPSERA with drugs that are excreted renally, or other drugs known to affect renal function have not been evaluated (See CLINICAL PHARMACOLOGY ).
Patients should be monitored closely for adverse events when HEPSERA is co-administered with drugs that are excreted renally or with other drugs known to affect renal function.
Ibuprofen 800 mg three times daily increased adefovir exposure by approximately 23%. The clinical significance of this increase in adefovir exposure is unknown (See CLINICAL PHARMACOLOGY ).
While adefovir does not inhibit common CYP450 enzymes, the potential for adefovir to induce CYP450 enzymes is not known.
The effect of adefovir on cyclosporine and tacrolimus concentrations is not known.
Duration of Treatment
The optimal duration of HEPSERA treatment and the relationship between treatment response and long-term outcomes such as hepatocellular carcinoma or decompensated cirrhosis are not known.
Animal Toxicology
Renal tubular nephropathy characterized by histological alterations and/or increases in BUN and serum creatinine was the primary dose-limiting toxicity associated with administration of adefovir dipivoxil in animals. Nephrotoxicity was observed in animals at systemic exposures approximately 3-10 times higher than those in humans at the recommended therapeutic dose of 10 mg/day.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies in mice and rats receiving adefovir have been conducted. In mice, at dose levels of 1, 3, or 10 mg/kg/day, no treatment-related increases in tumor incidence were found at 10 mg/kg/day (systemic exposure was 10 times that achieved in humans at a therapeutic dose of 10 mg/day). In rats dosed at levels of 0.5, 1.5, or 5 mg/kg/day, no drug-related increase in tumor incidence was observed. The exposure at the high dose was four times that at the human therapeutic dose. Adefovir dipivoxil was mutagenic in the in vitro mouse lymphoma cell assay (with or without metabolic activation). Adefovir induced chromosomal aberrations in the in vitro human peripheral blood lymphocyte assay without metabolic activation. Adefovir was not clastogenic in the in vivo mouse micronucleus assay at doses up to 2,000 mg/kg and it was not mutagenic in the Ames bacterial reverse mutation assay using S. typhimurium and E. coli strains in the presence and absence of metabolic activation. In reproductive toxicology studies, no evidence of impaired fertility was seen in male or female rats at doses up to 30 mg/kg/day (systemic exposure 19 times that achieved in humans at the therapeutic dose).
Pregnancy
Pregnancy Category C:
Reproduction studies conducted with adefovir dipivoxil administered orally have shown no embryotoxicity or teratogenicity in rats at doses up to 35 mg/kg/day (systemic exposure approximately 23 times that achieved in humans at the therapeutic dose of 10 mg/day), or in rabbits at 20 mg/kg/day (systemic exposure 40 times human).
When adefovir was administered intravenously to pregnant rats at doses associated with notable maternal toxicity (20 mg/kg/day, systemic exposure 38 times human), embryotoxicity and an increased incidence of fetal malformations (anasarca, depressed eye bulge, umbilical hernia and kinked tail) were observed. No adverse effects on development were seen with adefovir administered intravenously to pregnant rats at 2.5 mg/kg/day (systemic exposure 12 times human).
There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, HEPSERA should be used during pregnancy only if clearly needed and after careful consideration of the risks and benefits.
Pregnancy Registry
To monitor fetal outcomes of pregnant women exposed to HEPSERA, a pregnancy registry has been established. Healthcare providers are encouraged to register patients by calling 1-800-258-4263.
Labor and Delivery
There are no studies in pregnant women and no data on the effect of HEPSERA on transmission of HBV from mother to infant. Therefore, appropriate infant immunizations should be used to prevent neonatal acquisition of hepatitis B virus.
Lactating Women
It is not known whether adefovir is excreted in human milk. Mothers should be instructed not to breast-feed if they are taking HEPSERA.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Clinical studies of HEPSERA did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. In general, caution should be exercised when prescribing to elderly patients since they have greater frequency of decreased renal or cardiac function due to concomitant disease or other drug therapy.
ADVERSE REACTIONS
Assessment of adverse reactions is based on two studies (437 and 438) in which 522 patients with chronic hepatitis B received double-blind treatment with HEPSERA (n = 294) or placebo (n = 228) for 48 weeks. With extended therapy in the second 48 week treatment period, 492 patients were treated for up to 109 weeks, with a median time on treatment of 49 weeks.
In addition to specific adverse events described under the WARNINGS section, all treatment-related clinical adverse events that occurred in 3% or greater of HEPSERA-treated patients compared with placebo are listed in Table 6. A summary of grade 3 and 4 laboratory abnormalities during therapy with HEPSERA compared with placebo is listed in Table 7.
Table 6. Treatment-Related Adverse Events (Grades 1-4) Reported in >/= 3% of All HEPSERA-Treated Patients in the Pooled 437 - 438 Studies (0-48 Weeks)
| HEPSERA 10 mg | Placebo | |
| (n = 294) | (n = 228) | |
| Asthenia | 13% | 14% |
| Headache | 9% | 10% |
| Abdominal pain | 9% | 11% |
| Nausea | 5% | 8% |
| Flatulence | 4% | 4% |
| Diarrhea | 3% | 4% |
| Dyspepsia | 3% | 2% |
Laboratory Abnormalities
Table 7. Grade 3-4 Laboratory Abnormalities Reported in >/= 1% of All HEPSERA-Treated Patients in the Pooled 437 - 438 Studies (0-48 Weeks)
| HEPSERA 10 mg | Placebo | |
| (n = 294) | (n = 228) | |
| ALT (> 5 x ULN) | 20% | 41% |
| Hematuria (>/= 3+) | 11% | 10% |
| AST (> 5 x ULN) | 8% | 23% |
| Creatine Kinase (> 4 x ULN) | 7% | 7% |
| Amylase (> 2 x ULN) | 4% | 4% |
| Glycosuria (>/= 3+) | 1% | 3% |
In patients with adequate renal function, increases in serum creatinine >/= 0.3 mg/dL from baseline were observed in 4% of patients treated with HEPSERA 10 mg daily compared with 2% of patients in the placebo group by week 48. No patients developed a serum creatinine increase >/= 0.5 mg/dL from baseline by week 48. By week 96, 10% and 2% of HEPSERA-treated patients, by Kaplan-Meier estimate, had increases in serum creatinine >/= 0.3 mg/dL and >/= 0.5 mg/dL from baseline, respectively (no placebo-controlled results were available for comparison beyond week 48). Of the 29 of 492 patients with elevations in serum creatinine >/= 0.3 mg/dL from baseline, 20 out of 29 resolved on continued treatment (</= 0.2 mg/dL from baseline), 8 of 29 remained unchanged and 1 of 29 resolved on discontinuing treatment (See Special Risk Patients section below for changes in serum creatinine in patients with underlying renal insufficiency at baseline).
Special Risk Patients
Pre- (n = 128) and post-liver transplantation patients (n = 196) with chronic hepatitis B and clinical evidence of lamivudine-resistant hepatitis B virus were treated in an open-label study with HEPSERA for up to 129 weeks, with a median time on treatment of 19 and 56 weeks, respectively. The majority of these patients had some degree of underlying renal insufficiency at baseline or other risk factors for renal dysfunction during treatment. Increases in serum creatinine >/= 0.3 mg/dL from baseline were observed in 26% of these patients by week 48 and 37% by week 96 by Kaplan-Meier estimates. Increases in serum creatinine >/= 0.5 mg/dL from baseline were observed in 16% of these patients by week 48 and 31% by week 96. Of the 41 of 324 patients with elevations in serum creatinine >/= 0.5 mg/dL from baseline, 7 of 41 resolved on continued treatment (</= 0.3 mg/dL from baseline), 18 of 41 remained unchanged and 16 of 41 had not resolved. Additionally, deceases in serum phosphorus were observed in 4% of these patients by week 48, and 6% by week 96 by Kaplan-Meier estimates. One percent (3 of 324) of pre- and post-liver transplantation patients discontinued HEPSERA due to renal events.
Due to the presence of multiple concomitant risk factors for renal dysfunction in these patients, the contributory role of HEPSERA to these changes in serum creatinine and serum phosphorus is difficult to assess.
The most common treatment-related adverse events reported in pre- and post-liver transplantation patients treated with HEPSERA with a 2% frequency or higher include:
- Body as a whole: asthenia, abdominal pain, headache, fever
- Gastrointestinal: nausea, vomiting, diarrhea, flatulence, hepatic failure
- Metabolic and Nutritional: increases in ALT and AST, abnormal liver function
- Respiratory: increased cough, pharyngitis, sinusitis
- Skin and Appendages: pruritus, rash
- Urogenital: increases in creatinine, renal failure, renal insufficiency
OVERDOSAGE
Doses of adefovir dipivoxil 500 mg daily for 2 weeks and 250 mg daily for 12 weeks have been associated with gastrointestinal side effects. If overdose occurs the patient must be monitored for evidence of toxicity, and standard supportive treatment applied as necessary.
Following a 10 mg single dose of HEPSERA, a four-hour hemodialysis session removed approximately 35% of the adefovir dose.
DOSAGE AND ADMINISTRATION
The recommended dose of HEPSERA in chronic hepatitis B patients with adequate renal function is 10 mg, once daily, taken orally, without regard to food. The optimal duration of treatment is unknown.
Dose Adjustment in Renal Impairment
Significantly increased drug exposures were seen when HEPSERA was administered to patients with renal impairment (See Pharmacokinetics). Therefore, the dosing interval of HEPSERA should be adjusted in patients with baseline creatinine clearance < 50 mL/min using the following suggested guidelines (See Table 8). The safety and effectiveness of these dosing interval adjustment guidelines have not been clinically evaluated. Additionally, it is important to note that these guidelines were derived from data in patients with pre-existing renal impairment at baseline. They may not be appropriate for patients in whom renal insufficiency evolves during treatment with HEPSERA. Therefore, clinical response to treatment and renal function should be closely monitored in these patients.
Table 8. Dosing Interval Adjustment of HEPSERA in Patients with Renal Impairment
| Creatinine Clearance (mL/min) * | ||||
| >/= 50 | 20-49 | 10-19 | Hemodialysis | |
| Recommended Dose and Dosing Interval |
10 mg every 24 hours |
10 mg every 48 hours |
10 mg every 72 hours |
10 mg every 7 days following dialysis |
*Creatinine clearance calculated by Cockcroft-Gault method using lean or ideal body weight
The pharmacokinetics of adefovir have not been evaluated in non-hemodialysis patients with creatinine clearance < 10 mL/min; therefore, no dosing recommendation is available for these patients.
HOW SUPPLIED
HEPSERA is available as tablets. Each tablet contains 10 mg of adefovir dipivoxil. The tablets are white and debossed with "10" and "GILEAD" on one side and the stylized figure of a liver on the other side. They are packaged as follows: Bottles of 30 tablets (NDC 61958-0501-1) containing desiccant (silica gel) and closed with a child-resistant closure.
Store in original container at 25 °C (77 °F), excursions permitted to 15-30 °C (59-86 °F) (See USP Controlled Room Temperature).
Do not use if seal over bottle opening is broken or missing.
 Gilead Sciences, Inc.
Gilead Sciences, Inc.
Foster City, CA 94404
June 2003
HEPSERA™ is a trademark of Gilead Sciences
©2002,2003 Gilead Sciences, Inc.
GILEAD
PRODUCT PHOTO(S):
NOTE: These photos can be used only for identification by shape, color, and imprint. They do not depict actual or relative size.
Перевод этой статьи на русский язык можно посмотреть здесь.
Форма обратной связи
- О компании
- Качество
- Примеры переводов
- Карта сайта
- Нормативные документы
- Конфиденциальность
- Обработка персональных данных
- Ограничение ответственности
